

FDA Proposes New Approval Pathway for Ultra-Rare Disease Therapies
By Amy Sapola, Pharm.D., Functional Medicine Practitioner, Contributor, The MAHA Report
For years, families confronting ultra-rare genetic conditions have faced a painful truth: science may understand the mutation of a disease, but the regulatory pathway to an approved therapy simply did not exist. That may soon change.

On February 23, the U.S. Food and Drug Administration (FDA) announced a new regulatory framework designed to accelerate the development of individualized therapies for ultra-rare diseases. At the center of this effort is what the agency calls a “plausible mechanism” approach. This model allows approval decisions to rely more heavily on strong biological rationale and mechanistic evidence when traditional large clinical trials are not feasible.
There are an estimated 7,000 to 10,000 recognized rare diseases, affecting approximately 25 to 30 million Americans. About 80 percent of these conditions are genetic in origin, and roughly half present in childhood. This means nearly 1 in 10 American families are touched by a rare disease in some form. Ultra-rare diseases represent a smaller but deeply significant subset of that population. Many of these conditions affect fewer than 1,000 individuals in the United States, and some only a few dozen, making precise prevalence estimates difficult.
The paradox is striking: while a single ultra-rare disease may impact 5, 20, or 200 families, there are thousands of such diseases. Individually rare. Collectively common.
The FDA’s draft guidance outlines a pathway in which substantial evidence of effectiveness and safety can be supported by a clearly defined biological cause of disease, a therapy that directly targets that abnormality, and evidence that the therapy meaningfully modifies the disease pathway. In other words, if we can demonstrate that a genetic mutation causes a predictable dysfunction and we can show that the therapy corrects that dysfunction at a molecular or cellular level, that mechanistic clarity can serve as a core evidentiary pillar.
This approach represents regulatory alignment with modern molecular medicine. It is now possible to design therapies that target the exact mutation driving the disease. Yet the regulatory system has struggled to keep pace.
The plausible mechanism framework attempts to close that gap. It allows sponsors to integrate natural history data, mechanistic evidence, biomarker changes, and carefully collected clinical observations. Rather than dismissing small patient numbers as an insurmountable limitation, the guidance acknowledges that ultra-rare diseases require a different approach.
Importantly, this does not mean lowering safety standards. Individualized therapies, particularly gene and RNA-based approaches, carry significant scientific and ethical responsibility. The framework emphasizes early FDA engagement, careful manufacturing controls, and rigorous post-approval monitoring. In ultra-rare conditions, real-world evidence becomes not an afterthought, but an essential extension of the approval decision.

“This guidance is a critical step the FDA is taking to tailor our regulatory approach to patients with ultra-rare conditions,” said FDA Commissioner Marty Makary.“It is our priority to remove barriers and exercise regulatory flexibility to encourage scientific advances and deliver more cures and meaningful treatments for patients suffering from rare diseases.”
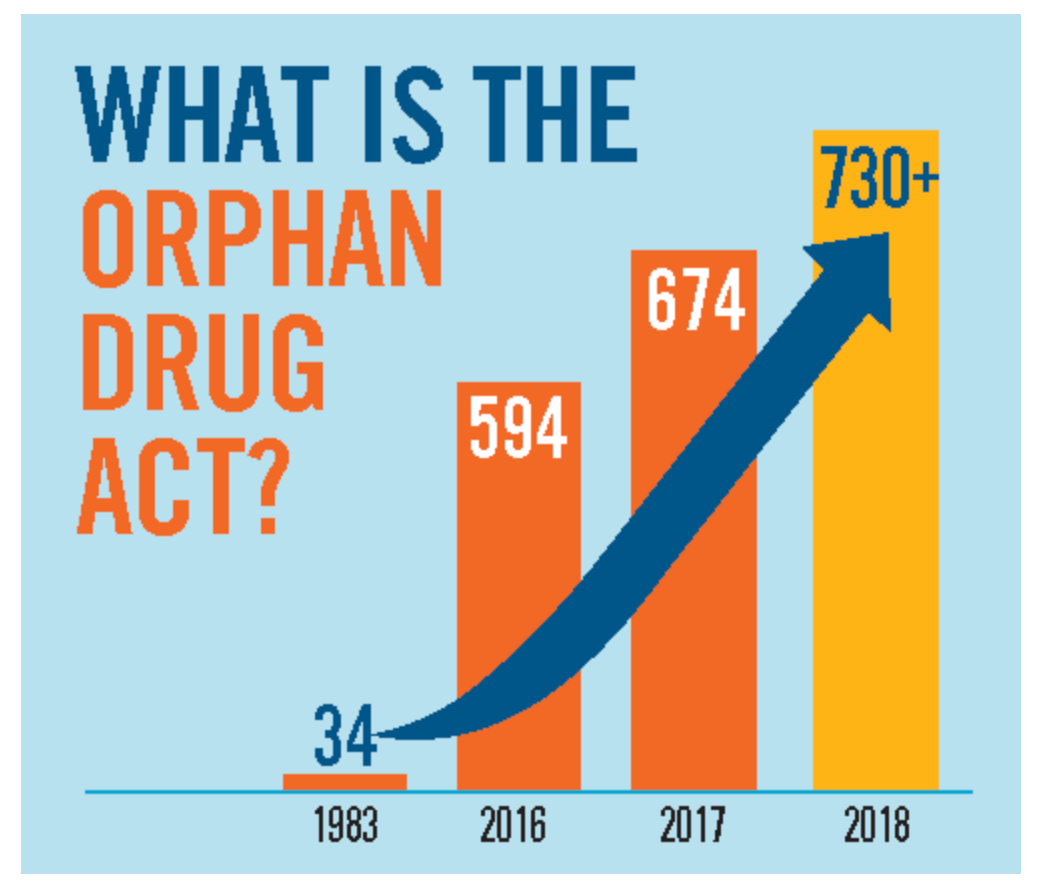
For families living in the margins of medicine, this is more than a policy update. The Orphan Drug Act of 1983 had already made rare disease drug development financially possible. The FDA’s new plausible mechanism framework makes ultra-rare, individualized therapy approval scientifically feasible.
Under conventional approval pathways, sponsors are expected to conduct randomized, adequately powered clinical trials to demonstrate safety and efficacy. But what happens when only a handful of patients exist globally? What happens when each patient carries a distinct pathogenic variant that requires a slightly different therapeutic construct?

“President Trump promised to accelerate cures for American families — and we are delivering, especially for children with ultra-rare diseases who cannot afford to wait,” said Health and Human Services Secretary Robert F. Kennedy Jr. “We are cutting unnecessary red tape, aligning regulation with modern biology, and clearing a path for breakthrough treatments to reach the patients who need them most.”
As a clinician, I see the FDA’s new guidance as part of a broader modernization of regulatory science. Medicine has moved decisively toward precision genomics, pharmacogenomics, and individualized risk stratification, yet regulatory paradigms have remained rooted in population averages. Ultra-rare disease forces us to confront the limits of that model.
The guidance also raises important policy questions. How do we ensure equitable access to individualized therapies that may be extraordinarily costly to develop? How do payers evaluate value when evidence comes from small, mechanistically justified datasets rather than large randomized trials? How do we balance accelerated approval with long-term safety surveillance?
These questions deserve rigorous debate. But they should not obscure the central achievement: the FDA is signaling that patients with ultra-rare diseases are not invisible to the system. The agency is articulating a pathway that recognizes scientific plausibility, mechanistic coherence, and biological causality as meaningful forms of evidence when traditional trials are impossible.
For families living with progressive genetic conditions, time is not an abstract concept. It is measured in lost milestones, declining function, and narrowing windows for intervention. A framework that compresses regulatory timelines while preserving scientific integrity saves valuable time that could translate into preserved function and extended life.
The FDA’s draft guidance is open for public comment. Patient advocacy groups, clinicians, trial methodologists, and sponsors have an opportunity to shape how this framework is finalized and implemented.
Ultra-rare disease drug development has long operated in a gray zone between scientific possibility and regulatory impracticality. With the plausible mechanism framework, that gray zone begins to resolve into a clearer path.
For decades, families confronting these diagnoses have been told that their numbers are too small. This framework suggests something different: that rarity does not negate worth, and that regulatory science can evolve to meet the frontiers of molecular medicine.
"Rarity does not negate worth." Says it all.
From US Right to Know newsletter
The White House invokes the Defense Production Act to guarantee supplies of elemental phosphorus and glyphosate-based herbicides. Regulators reapprove dicamba, a Bayer herbicide twice blocked by federal courts, and clear the way for new pesticides containing toxic, persistent PFAS “forever” chemicals.
And the U.S. Justice Department urges the Supreme Court to erase billions of dollars of Bayer’s liability for its glyphosate-based Roundup – placing the weight of the executive branch on the side of a foreign company against thousands of Americans who say Bayer’s products caused their cancers.
Over the past year, the Trump administration has delivered a string of victories to Bayer, the German agrichemical and pharmaceutical giant that merged with Monsanto in 2018 to become the world’s leading manufacturer of genetically modified seeds and pesticides.
What is Bayer’s access in Washington? Our review found 16 key administration officials with ties to Bayer’s lobbying or legal network. Bayer and its lobbyists have access to people in power at the White House, U.S. Department of Agriculture, the Environmental Protection Agency and even those in high-level positions closest to Trump.
Read Stacy Malkan’s reporting, Tracing Bayer’s ties to power in Trump’s Washington; From lobby firms to top officials, a look at how Bayer built access and secured favors
The most potent Bayer-Trump connections involve a group of Florida lobbyists and former lobbyists who hold power in Trump’s Washington: Chief of Staff Susie Wiles (left), Attorney General Pam Bondi (right), and Brian Ballard (middle), who employed both Bondi and Wiles as lobbyists for years.
Donate to Support our Right to Know
We are also tracking Bayer’s formidable lobbying force in Washington. As of the fourth quarter of 2025, the company retained 45 registered lobbyists and at least 13 outside lobby firms – seven of which are now among the highest-paid firms in D.C.
More than 30 senior officials at lobby firms retained by Bayer have direct ties to Trump, having worked in one or both of his administrations or political campaigns